Làm thế nào để một thiết bị thẩm mỹ nói riêng hay thiết bị y tế nói chung được FDA chứng nhận.
Quy trình chứng nhận như thế nào ? Mời các bạn tham khảo bài viết sau đây.
FFDCA = Luật Thực phẩm, Ma túy và Mỹ phẩm Liên bang Mỹ
HDE = Miễn trừ kiểm định thiết bị lâm sàng trên con người;
IDE = Miễn trừ kiểm định thiết bị phòng nghiên cứu;
IRB = Ủy Ban Xét duyệt
PMA = Phê duyệt trước khi ra thị trường;
HUD = Thiết bị sử dụng cho mục đích nhân đạo.
PMN = Thông báo trước khi ra thị trườngHU510 (k) là một bản đệ trình trước khi vào thị trường được thực hiện lên FDA để chứng minh rằng thiết bị được bán trên thị trường ít nhất cũng phải an toàn và hiệu quả, nghĩa là tương đương với một thiết bị xuất hiện trên thị trường hợp pháp.

Bước 1: Phân loại thiết bị
Bước đầu tiên cho người đăng ký (sau khi xác định sản phẩm là thiết bị) là phân loại thiết bị loại I, II, hoặc III.
Mặc dù FDA vẫn sẽ thực hiện điều này trong quá trình xem xét việc đệ trình nhưng điều quan trọng là người đăng ký cũng phải phân loại trước, bởi vì nó sẽ xác định những bước tiếp theo phù hợp với thiết bị.
Để hỗ trợ, FDA cung cấp một cơ sở dữ liệu online để tìm kiếm cách phân loại thiết bị . Phân loại của thiết bị này cũng sẽ xác định việc kiểm soát quy định trong và sau quá trình đăng ký.
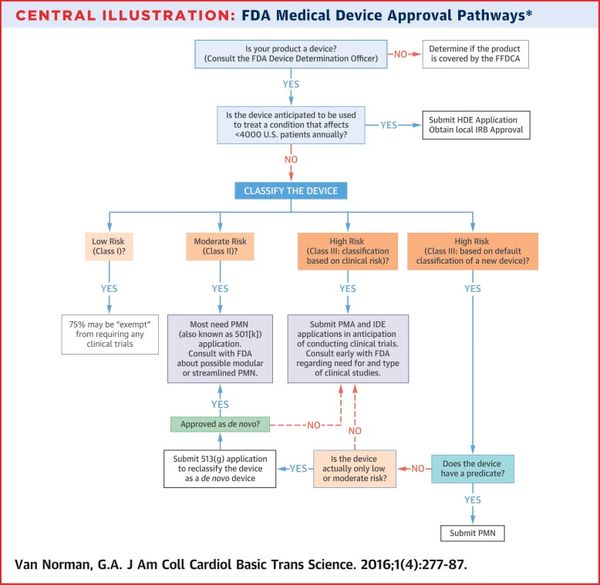
Bước 2: Chọn các bước đăng ký thích hợp.
Các thiết bị thuộc lớp I và nhiều thiết bị lớp II - lớp III có thể bỏ qua PMA và tiến hành 510 (k).
Phải có PMA cho hầu hết các thiết bị lớp III, có bằng chứng khoa học ở Mức I hoặc II hợp lệ cung cấp sự đảm bảo hợp lý về tính an toàn và hiệu quả.
Nếu thiết bị này là loại III và có lợi cho ít hơn 4.000 bệnh nhân hàng năm ở Hoa Kỳ, điều tra viên nên tiến hành yêu cầu chỉ định thiết bị như là một HUD. Sau khi nhận được chỉ định như là một HUD, điều tra viên nên chuẩn bị một ứng dụng HDE để miễn thiết bị không phải cung cấp bằng chứng nghiêm ngặt về hiệu quả.
Bước 3: Nếu thiết bị cần một PMA đầy đủ
Các thử nghiệm lâm sàng sẽ được yêu cầu và điều tra viên nên chuẩn bị một ứng dụng IDE. PMA có thể tiến hành sau khi có đủ chứng cứ lâm sàng cấp I và II.
Bước 4: Chuẩn bị ứng dụng phù hợp
FDA cung cấp thông tin tại trang web của họ để hỗ trợ các nhà đăng ký về các thông tin cần thiết, tùy thuộc vào việc ứng dụng đó là PMA, PMN hay HDE.
Nói chung, người nộp đơn phải cung cấp thông tin về các chỉ dẫn và chức năng của thiết bị, các khái niệm khoa học cơ bản, tóm tắt các thông tin về hiệu quả và an toàn - bất lợi, các quy trình kiểm soát rủi ro, các thủ tục và phương pháp điều trị thay thế đối với thiết bị, bản tóm tắt về tiền lâm sàng và (trong trường hợp của PMA, HDE và khoảng 501 [k]) dữ liệu thử nghiệm lâm sàng và một tài liệu tham khảo về tài liệu tham khảo.
Liên lạc trước khi nộp đơn với FDA để có phản hồi về việc phê duyệt thiết bị y tế.
Trước khi bắt đầu bất kỳ nghiên cứu lâm sàng nào của thiết bị cho mục đích PMA, HDE, hoặc 501 (k), điều tra viên phải có được IDE từ FDA cũng như sự chấp thuận của IRB địa phương.
Bước 5: Gửi bản đệ trình lên FDA
Hãy nhớ rằng sẽ có một khoản phí cho người dùng. Trong vòng 15 ngày kể từ ngày nhận được bản đệ trình, FDA sẽ hoàn thành việc xem xét chấp nhận hành chính và trả lời.
Nếu có vấn đề liên quan đến lệ phí người sử dụng, eCopy, hoặc thông tin trong gói, việc xem xét có thể dẫn đến "treo", và người đăng ký sẽ được thông báo. Sau đó có 180 ngày để giải quyết các vấn đề. Nếu không có vấn đề gì (hoặc sau khi các vấn đề được giải quyết), FDA sẽ chỉ định một nhà phê duyệt chính và thông báo cho người nộp đơn về sự chấp nhận để được xem xét hoàn chỉnh.
Thông tin liên lạc của FDA với người nộp đơn sẽ thường xuyên trong khi thiết bị đang được xem xét để tăng hiệu quả của quá trình xem xét.
FDA chỉ định thời gian tương tác xảy ra như sau:
1) trong vòng 60 ngày kể từ ngày nhận được bản đệ trình đầy đủ cho 510 (k) (tức là 45 ngày sau khi chấp nhận để xem xét hoàn chỉnh)
2) trong vòng 90 ngày kể từ ngày nộp đơn cho PMA (tức là, trong vòng 75 ngày kể từ ngày nhận được để xem xét hoàn chỉnh).
Bước 6: Đăng ký thành lập và liệt kê thiết bị
Sau khi FDA chấp thuận, người đăng ký phải đăng ký kinh doanh sản xuất và phân phối thiết bị bên trong Hoa Kỳ và "liệt kê" thiết bị của họ.
Kinh doanh: Mr. Khánh 0909002319 - Ms. Thoa 0907533288
Kỹ thuật : Mr. Di 0939479932
Email: amajsc2017@gmail.com